If you're working in a multiple-compiler environment, you may wish to get information at compile time about which compiler you're building with. Here's a list of macros defined by many compilers:
https://sourceforge.net/p/predef/wiki/Compilers/
Thursday, February 27, 2020
Friday, February 14, 2020
Enabling wind power nationwide
A report from the Obama Department of Energy in 2015.
https://www.energy.gov/sites/prod/files/2015/05/f22/Enabling%20Wind%20Power%20Nationwide_18MAY2015_FINAL.pdf
https://www.energy.gov/sites/prod/files/2015/05/f22/Enabling%20Wind%20Power%20Nationwide_18MAY2015_FINAL.pdf
Thursday, February 13, 2020
Take a newick tree and stre-e-etch the leaves to make it ultrametric
I needed to generate some ultrametric Newick trees for some simulations. There's a nice Newick tree generator online at http:/Trex/trex.uqam.ca/index.php?action=randomtreegenerator&project=trex, but it doesn't have any way to make an ultrametric tree. (Ultrametric means that the lengths from root to tip are all of the same.) So I put together a short script using BioPython to replace the length of each leaf node to make the lengths all identical to the longest length.
How to delete empty rows in an Excel spreadsheet
How to delete empty rows in an Excel spreadsheet:
https://www.itsupportguides.com/knowledge-base/office-2016/excel-2016-how-to-delete-empty-rows/
https://www.itsupportguides.com/knowledge-base/office-2016/excel-2016-how-to-delete-empty-rows/
Monday, August 10, 2015
How Installing a Random Tool Caused Ruby on Rails to Stop Working
At work I maintain a little Ruby on Rails site. Not a particularly interesting site, just a set of pages of statistics gathered from various databases around the organization. It runs on a RHEL6 virtual machine and I write most of the code on my Windows laptop using RubyMine from JetBrains - a product I wholeheartedly endorse, by the way.
It does take a little patience to run Rails on Windows - not due to any deficit in either product, just because the vast majority of people who work with Rails are running it on some other platform, so when you have problems, a lot of times you're on your own.
Take this problem I ran into recently, for example. I launched RubyMine to check out a bug, fired up the ol' testing server, browsed to the web page, and was greeted with this:
JSON::ParserError in Statistics#index
Showing /app/views/layouts/application.html.erb where line #5 raised:
757: unexpected token at 'Node Commands
Syntax:
node {operator} [options] [arguments]
Parameters:
/? or /help - Display this help message.
list - List nodes or node history or the cluster
listcores - List cores on the cluster
view - View properties of a node
online - Set nodes or node to online state
offline - Set one or more nodes to the offline state
For more information about HPC command-line tools,
see http://go.microsoft.com/fwlink/?LinkId=120724.
(in /app/assets/javascripts/accounts.js.coffee)
Now, of all possible error messages to get, one involving HPC command-line tools was not one that I had ever considered. What's going on here?
The line that gives the error is one that occurs in every Rails application:
It does take a little patience to run Rails on Windows - not due to any deficit in either product, just because the vast majority of people who work with Rails are running it on some other platform, so when you have problems, a lot of times you're on your own.
Take this problem I ran into recently, for example. I launched RubyMine to check out a bug, fired up the ol' testing server, browsed to the web page, and was greeted with this:
JSON::ParserError in Statistics#index
Showing /app/views/layouts/application.html.erb where line #5 raised:
757: unexpected token at 'Node Commands
Syntax:
node {operator} [options] [arguments]
Parameters:
/? or /help - Display this help message.
list - List nodes or node history or the cluster
listcores - List cores on the cluster
view - View properties of a node
online - Set nodes or node to online state
offline - Set one or more nodes to the offline state
For more information about HPC command-line tools,
see http://go.microsoft.com/fwlink/?LinkId=120724.
(in /app/assets/javascripts/accounts.js.coffee)
Now, of all possible error messages to get, one involving HPC command-line tools was not one that I had ever considered. What's going on here?
The line that gives the error is one that occurs in every Rails application:
<%= javascript_include_tag 'application' %>
What it really comes down to saying is, take all of the Javascript and Coffeescript code in the application, and include it here. A little wasteful, perhaps, but in a production environment all of the code will get glommed into a single file appropriate for caching in the browser, so it's not a bad system. Still, where's the error?
Obviously it must be in the Coffeescript file referenced at the end of the error message. But why is it suddenly complaining about HPC? I went and commented that whole file out - no joy. The same error got reported with a different file. I could find no information anywhere about what the cause might have been, and I didn't expect searching for the error message itself would do any good. I work in an HPC - which stands for High Performance Computing, incidentally - environment so I was fairly sure the error was something very specific to my machine.
I tried, of course. Several different Google searches failed - but I finally hit on the right combination of keywords that led me to a Google Plus comment on a YouTube tutorial.
It turns out that Rails, in its infinite struggle for flexibility, relies on a gem called ExecJS to decide how it's going to convert Coffeescript to Javascript. ExecJS, in turn, is of the opinion that Node.js is a really good way to do the conversion. And it determines if Node.js is available by trying to run an application called Node.
Which is fine, unless you happen to run Windows; and you happen to work in an HPC environment; and you happen to have gone to a conference the other week where you decided to install Microsoft's HPC tools; which happen to include a nice command for managing compute nodes; called - you guessed it - Node. ExecJS was trying to use Microsoft's Node command to convert Coffeescript to Javascript, resulting in the error you see. Hats off to Jonathan McDonald for nailing the issue.
Wednesday, July 16, 2014
Trinity RNA-Seq
In January a site went live called readingroom.info. I suspect due to the timing and the subject matter it was a student project. The idea was to write summaries - no more than 500 words - of scientific papers and allow people to comment on and discuss them. I thought it was a neat idea. They had some ideas for incentivizing writers and so forth, but I didn't have time to contribute anything until this summer, by which time the authors had apparently lost interest. I sent in a summary of a paper, but after several weeks it had not been approved by the moderators. Maybe it just wasn't that good!
The paper is Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data.
This is what I sent them:
The paper is Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data.
This is what I sent them:
The reconstruction of a
transcriptome from the
short reads generated by RNA-Seq techniques presents
many challenges, particularly in the absence of an existing reference genome
with which to compare the reads. Challenges include: uneven coverage of the
various transcripts; (ii) uneven coverage inside each transcript; (iii)
sequencing errors in highly expressed transcripts; (iv) transcripts encoded by
adjacent loci can overlap and thus can be erroneously fused to form a chimeric transcript; (v)
data structures need to accommodate multiple transcripts per locus, owing to
alternative splicing; and (vi) sequences that are repeated in different genes
introduce ambiguity. The Trinity pipeline leverages several properties of
transcriptomes in its assembly procedure: it uses transcript expression to
guide the initial transcript assembly procedure in a strand-specific manner, it
partitions RNA-Seq reads into sets of disjoint transcriptional loci, and it traverses
each of the transcript graphs systematically to explore the sets of transcript
sequences that best represent variants resulting from alternative splicing or
gene duplication by exploiting pairs of RNA-Seq reads. The series of steps
performed by the pipeline correctly reconstructs a significant percentage of
the transcripts without relying on the existence of a reference genome.
A major data structure used the
pipeline is the de Bruijn graph. A de Bruijn graph places each
k-mer in a node, and has
connected nodes if the k-mers are identical in all but the first or last
position. While an efficient structure for representing heavily overlapping
sequences, there are challenges in the usage of these graphs: (i) efficiently
constructing this graph from large amounts (billions of base pairs) of raw
data; (ii) defining a suitable scoring and enumeration algorithm to recover all
plausible splice forms and paralogous transcripts; and (iii) providing
robustness to the noise stemming from sequencing errors and other artifacts in
the data. Sequencing errors would introduce false nodes to the graph,
potentially resulting in a great deal of wasted memory.
The Trinity pipeline consists of
the following steps: it first analyzes the short reads to create a dictionary
of all sequences of length 25 in the reads, indexing the locations where each
sequence may be found. After removing likely errors, the unique k-mers are recombined,
starting with the most frequently occurring sequences and extending the
combination until no more k-mers can be matched. Each contig is then added to a
cluster based on potential alternative spliced transcripts or otherwise unique
portions of paralogous genes. Then, a de Bruijn graph is generated from each
cluster with the weight of each edge assigned from the number of k-mers in the
original read set that support the connection. In the final phase, a
merge-and-prune operation on each graph, for error correction, is performed,
followed by an enumeration of potential paths through the graph with a greater
likelihood placed on paths with greater read support.
The authors built transcriptomes
from both original data and reference sets, having a great deal of success in
either case.
Tuesday, July 08, 2014
The Slim protocol
I wrote earlier about Fitnesse and Slim style integration testing.
There are only a few commands associated with Slim tables, but the precise meaning of the command is left to the Slim server. For example, according to the Slim documentation, the Import instruction:
causes the <path> to be added to the search path for fixtures. In java <path> gets added to the CLASSPATH. In .NET, the <path> would name a dll.
This explains why import tests are always green. You can pass them any random string and it's just added to the path. In WaferSlim, the concept is extended slightly: if the string has a slash or backslash in it, it's considered a path. Otherwise, it's considered a file and is added to the list of files searched for classes. Let's take an example. Create a file called fixtures.py with the following code:
class SomeDecisionTable:
def setInput(self, x):
self.x = str(x)
def getOutput(self):
return int(self.x) + 1
Make: SomeDecisionTable
Call: setInput(1)
Call: getOutput()
Call: setInput(10)
Call: getOutput()
Click the test button now and everything should run green.
There are only a few commands associated with Slim tables, but the precise meaning of the command is left to the Slim server. For example, according to the Slim documentation, the Import instruction:
causes the <path> to be added to the search path for fixtures. In java <path> gets added to the CLASSPATH. In .NET, the <path> would name a dll.
This explains why import tests are always green. You can pass them any random string and it's just added to the path. In WaferSlim, the concept is extended slightly: if the string has a slash or backslash in it, it's considered a path. Otherwise, it's considered a file and is added to the list of files searched for classes. Let's take an example. Create a file called fixtures.py with the following code:
class SomeDecisionTable:
def setInput(self, x):
self.x = str(x)
def getOutput(self):
return int(self.x) + 1
There's an odd Unicode issue that I think requires context variables to be saved as strings. I don't know if Fitnesse, Slim or Waferslim is responsible for that. At any rate, it's simple enough: set the input to an integer and the output will be the integer plus one.
Save that file. Now the Import table will take two lines, one with the path, one with the file:
!|import |
|/path/to/fixtures|
|fixtures|
Now WaferSlim knows where to find our code, so let's take a look at another table.
Here's what the Slim protocol tells us: it can send Make instructions and Call instructions. A Make instruction tells the server to create an object; a Call instruction tells it to call a method on that object. I'd hoped to go through the Fitnesse code to determine exactly how that works, but I didn't. We'll take it on faith that the first line of a table sends a Make instruction and further lines send Call instructions. So, in order to make calls into our object, we write:
!|SomeDecisionTable|
|input|get output?|
|1 |2 |
!|SomeDecisionTable|
|input|get output?|
|1 |2 |
|10 |11 |
From the first line of the table, Fitnesse sends a Make: SomeDecisionTable command to the Slim server. Because of our Import statements, the server searches the /path/to/fixtures directory for the fixtures.py file, finds the SomeDecisionTable class, and instantiates it.
The second line of the table tells Fitnesse what Call statements to make. It will call setInput and then getOutput. Each further line of the table gives arguments for the input and output. So the sequence of events from the Slim server's perspective is as follows:
Make: SomeDecisionTable
Call: setInput(1)
Call: getOutput()
Call: setInput(10)
Call: getOutput()
Sunday, July 06, 2014
Fitnesse and Slim
Fitnesse is a rather nice testing tool. It's been around since what seems like the beginning of the integration testing movement. It's based around HTML tables that are translated into application code. The wiring that translates tables into code are called fixtures. Since Fitnesse was written in Java, it was mostly useful for testing Java code, although many translations and tricks were devised to allow tests of other languages. Fitnesse also includes a wiki to make the process of creating the test tables easier.
A few years after Fitnesse was introduced, an alternate translation tool, SLIM, was created. The idea behind Slim was to allow testers to implement tests in their language of choice, with the communication between Fitnesse and the tests taking place over a socket. This allows any application that implements the correct protocol to run tests written in Fitnesse tables.
Several applications were created to support the protocol in various languages, including RubySlim for Ruby and WaferSlim for Python. I had occasion to write a set of integration tests recently, so I thought it would be simple to set up a Slim server to get the tests running. Turned out, I was wrong.
For my tests, I wanted to set up the simplest thing that could possibly work. So what is the minimum requirement for a Slim test page? If you look at the RubySlim documentation you end up with a page that looks something like this:
What all these things do is not clearly explained. Click the test button; you get a bunch of cryptic error messages. So what do they mean?
Well, the first line is pretty clear: if you want to use Slim, you need to set the test system. For the rest of it, we pretty much need to go directly to the source code. It turns out that the heart of Slim is in the Java class ProcessBuilder. In the CommandRunner class of Fitnesse we find:
ProcessBuilder processBuilder = new ProcessBuilder(command);
The command argument is no more than the COMMAND_PATTERN variable defined on the page, with a single argument appended, a port number. So if you wanted to use WaferSlim (a Python Slim server), you might say:
!define COMMAND PATTERN {python /home/benfulton/slim-init.py}
A few years after Fitnesse was introduced, an alternate translation tool, SLIM, was created. The idea behind Slim was to allow testers to implement tests in their language of choice, with the communication between Fitnesse and the tests taking place over a socket. This allows any application that implements the correct protocol to run tests written in Fitnesse tables.
Several applications were created to support the protocol in various languages, including RubySlim for Ruby and WaferSlim for Python. I had occasion to write a set of integration tests recently, so I thought it would be simple to set up a Slim server to get the tests running. Turned out, I was wrong.
For my tests, I wanted to set up the simplest thing that could possibly work. So what is the minimum requirement for a Slim test page? If you look at the RubySlim documentation you end up with a page that looks something like this:
!define TEST_SYSTEM {slim}
!define TEST_RUNNER {rubyslim}
!define COMMAND_PATTERN {rubyslim}
!path your/ruby/fixtures
!|import|
|<ruby module of fixtures>|
|SomeDecisionTable|
|input|get output?|
|1 |2 |
What all these things do is not clearly explained. Click the test button; you get a bunch of cryptic error messages. So what do they mean?
Well, the first line is pretty clear: if you want to use Slim, you need to set the test system. For the rest of it, we pretty much need to go directly to the source code. It turns out that the heart of Slim is in the Java class ProcessBuilder. In the CommandRunner class of Fitnesse we find:
ProcessBuilder processBuilder = new ProcessBuilder(command);
The command argument is no more than the COMMAND_PATTERN variable defined on the page, with a single argument appended, a port number. So if you wanted to use WaferSlim (a Python Slim server), you might say:
!define COMMAND PATTERN {python /home/benfulton/slim-init.py}
(Since we need to understand how Slim works, let's get the WaferSlim source from Github rather than installing it via Pip. The slim-init source is from this gist.)
So, after downloading WaferSlim and the code from the gist, we should be able to get the Simplest Possible Page to work. Here it is:
!define TEST_SYSTEM {slim}
!define COMMAND_PATTERN {python /home/benfulton/slim-init.py }
!|import |
|RubySlim|
|app|
Click the test button, and you should get a green test.
If you don't, you may have a Slim versioning error. Go to protocol.py and change the version number from 0.1 to 0.3. Or Python may not be in your path, or the init-script may not be found. Or - here's a tricky one - you put a space before the python command. If you did that, the first command that gets passed to the ProcessBuilder class will be an empty string, and it won't run. Still, this will run green without too much effort.
But wait! What's the use of this import statement that went green before we wrote anything?
Sunday, June 02, 2013
Suffix Arrays
In our quest to win a million dollars, we're trying to Index the Human Genome. A typical index, like in the back of a book, is composed of an entry, say "Sheep", and a list of page numbers, say "88,121,265". But when we went to try this idea on the 3,000,000,000 characters in the genome, we found that it took up significantly more space than the genome itself, which was unworkable. Surely we can find a more efficient solution to the problem. (Or, we could throw up our hands, call it "Big Data", and put a supercomputer to work on it. Let's not do that.)
 Actually, there's a fair amount of redundancy in an index. If there are three instances of the word "Sheep" in our book, and we add a fourth instance of it to the index, we're increased the number of sheep, so to speak, by 33%. If we do that for every word in the book we'll add a significant chunk to the overall size.
Actually, there's a fair amount of redundancy in an index. If there are three instances of the word "Sheep" in our book, and we add a fourth instance of it to the index, we're increased the number of sheep, so to speak, by 33%. If we do that for every word in the book we'll add a significant chunk to the overall size.
But suppose, rather than just give the user the page numbers where the word "Sheep" is, we provided them with with the line and word numbers as well. "Sheep" is on page 88, line 14, word six. Now, the index is in alphabetical order of course, so what we can do is simply eliminate all those redundant words from the index. So the index entry for "Sheep" would just say "88,14,6". Say readers want to find the word "Streams". They would go and find an entry "88,14,6" and look up the word in the book. Finding that it's "Sheep", they realize the word is later in the index, since "Streams" is alphabetically after "Sheep". They go to the next entry in the index, maybe "81,19,11", and look up that word in the book. It's "Streams", so they've found their word, and the index didn't require any of those annoying, redundant words in it!
OK, not a very simple operation for a human reader. But easy enough for a computer. And since our genome doesn't have pages or lines, we could simply record the location of each individual 20-mer and put the locations in alphabetical order. We can even take it one step further: since 20 is just an arbitrary length that we chose, we'll remove it from the solution and just say that we'll take as many characters as we need to get a unique ordering of all of the strings. Notice that you might need to take all of the remaining characters of the genome to alphabetize it correctly, and if you do, you have a suffix of the genome. If you don't need all the characters, it doesn't matter if you add them or not, so you might as well, and therefore we have an array of the suffixes of the genome. A suffix array.
I won't go into the details of how to create such an index right now, but it can be done in relatively few lines of code. (One easy-to-use library is called SAIS.) Now, the simplest way to write out a suffix array is a text file of the alphabetized suffix indices, in ASCII format, one number per line. This, unfortunately, brings us straight back to the size problem - let's say eight bytes on average, per line, with one line per character in the genome, and we end up with 24 gigabytes worth of index. But at least it's a workable index. If we split the index into several files to keep it manageable, it even suggests a refinement of our attack on the overall problem. We'll see how another time.
(Update: I wrote an article on suffix array algorithms.)
Part I: A Million Dollars Up For Grabs
Part II: Analyzing DNA with BWA
Part III: Analyzing DNA Programmatically
Part IV: Indexing the Human Genome
Part V: Suffix Arrays
 Actually, there's a fair amount of redundancy in an index. If there are three instances of the word "Sheep" in our book, and we add a fourth instance of it to the index, we're increased the number of sheep, so to speak, by 33%. If we do that for every word in the book we'll add a significant chunk to the overall size.
Actually, there's a fair amount of redundancy in an index. If there are three instances of the word "Sheep" in our book, and we add a fourth instance of it to the index, we're increased the number of sheep, so to speak, by 33%. If we do that for every word in the book we'll add a significant chunk to the overall size.But suppose, rather than just give the user the page numbers where the word "Sheep" is, we provided them with with the line and word numbers as well. "Sheep" is on page 88, line 14, word six. Now, the index is in alphabetical order of course, so what we can do is simply eliminate all those redundant words from the index. So the index entry for "Sheep" would just say "88,14,6". Say readers want to find the word "Streams". They would go and find an entry "88,14,6" and look up the word in the book. Finding that it's "Sheep", they realize the word is later in the index, since "Streams" is alphabetically after "Sheep". They go to the next entry in the index, maybe "81,19,11", and look up that word in the book. It's "Streams", so they've found their word, and the index didn't require any of those annoying, redundant words in it!
 |
| Sheep. By a stream. |
I won't go into the details of how to create such an index right now, but it can be done in relatively few lines of code. (One easy-to-use library is called SAIS.) Now, the simplest way to write out a suffix array is a text file of the alphabetized suffix indices, in ASCII format, one number per line. This, unfortunately, brings us straight back to the size problem - let's say eight bytes on average, per line, with one line per character in the genome, and we end up with 24 gigabytes worth of index. But at least it's a workable index. If we split the index into several files to keep it manageable, it even suggests a refinement of our attack on the overall problem. We'll see how another time.
(Update: I wrote an article on suffix array algorithms.)
Part I: A Million Dollars Up For Grabs
Part II: Analyzing DNA with BWA
Part III: Analyzing DNA Programmatically
Part IV: Indexing the Human Genome
Part V: Suffix Arrays
Sunday, May 19, 2013
Indexing the human genome
Last time I had decided that to efficiently analyze the reads, we had to make an index of the human genome. So how do we go about that?

Indexing at the Library of Congress
What we need is an efficient way to access any given substring in the genome. It's not quite the same as indexing a book; rather than determining the locations of "dynamo", "father", and "pseudopodia" in the book, we need to be able to find the location of EVERY substring. It's as if, in our book, we had to find instances of "dynamo", "ynamo", "namo" and so on. Not only that, but if the book had the sentence, "A dynamo has unlimited duration." we have to find instances of "namo h", "namo ha" "namo has" and so on.
So we can't just split the genome by word boundaries like we would for a book. Can we split it into even-sized chunks and index those? For example, could we choose a chunk size of five and split every ten characters into two index entries?
It won't work. For example, if the genome was "AGACTTGCTG", we might choose to index every five characters (called a 5-mer). This would give us two strings, "AGACT" and "TGCTG", which is fine, but if we come along later and try to search for "CTTGC", we're out of luck - that's not in our index. But it is in the string.
So we have to choose an index size and go through the genome character by character. In our ten-character genome, indexing by 5-mer, we get the strings:
AGACT
GACTT
ACTTG
CTTGC
TTGCT
TGCTG
(and a few shorter strings at the end, if we so desire.)
To be useful, we'll have to store our index as a dictionary of 5-mers to an array of integers, representing the locations in the genome where that string was found. For our sample, we have 30 characters worth of 5-mers, and just six integers to save, for a total of 54 bytes. What happens when we index the whole genome?
It's a fair assumption that every possible string will be in the genome, so we'll have 45 or 1024 entries in our index. The number of values, though...we have to have an index value for every single character in the genome. If our genome was the book Moby Dick, we'd have around a million characters to index. Each index value would have, on average, about 1000 items in it. If we're really hoping to match all of our reads to each item in the index, we're going to go through 1000 entries for each read, which might be a bit slow. Unfortunately, our genome isn't Moby Dick. It's roughly the size of 6,000 Moby Dicks. Each index is going to be in just about six million locations. It can't possibly read all those at the speed we require.
Okay, so if we have too many items per index value, we just have to make our index larger. What if we do 10-mers, or even 20-mers? Well, 10-mers gives us 410 entries - call it a million. That means each index will be found in about 6,000 locations. Kind of a lot, but maybe doable. If we do 20-mers - well, 420 is 1,099,511,627,776. This is more on the lines of what we need in an index - it's well beyond the number of characters in the genome, so each index shouldn't show up in more than one or two locations. There's just one small problem: We now have six billion entries with twenty-character identifiers, and the space our index needs is now up to 120 gigabytes!
Maybe we could tweak and tune and find a sweet spot, but instead we'll try a different approach entirely to indexing. Next time.
Part I: A Million Dollars Up For Grabs
Part II: Analyzing DNA with BWA
Part III: Analyzing DNA Programmatically
Part IV: Indexing the Human Genome
Indexing at the Library of Congress
So we can't just split the genome by word boundaries like we would for a book. Can we split it into even-sized chunks and index those? For example, could we choose a chunk size of five and split every ten characters into two index entries?
It won't work. For example, if the genome was "AGACTTGCTG", we might choose to index every five characters (called a 5-mer). This would give us two strings, "AGACT" and "TGCTG", which is fine, but if we come along later and try to search for "CTTGC", we're out of luck - that's not in our index. But it is in the string.
So we have to choose an index size and go through the genome character by character. In our ten-character genome, indexing by 5-mer, we get the strings:
AGACT
GACTT
ACTTG
CTTGC
TTGCT
TGCTG
(and a few shorter strings at the end, if we so desire.)
To be useful, we'll have to store our index as a dictionary of 5-mers to an array of integers, representing the locations in the genome where that string was found. For our sample, we have 30 characters worth of 5-mers, and just six integers to save, for a total of 54 bytes. What happens when we index the whole genome?
It's a fair assumption that every possible string will be in the genome, so we'll have 45 or 1024 entries in our index. The number of values, though...we have to have an index value for every single character in the genome. If our genome was the book Moby Dick, we'd have around a million characters to index. Each index value would have, on average, about 1000 items in it. If we're really hoping to match all of our reads to each item in the index, we're going to go through 1000 entries for each read, which might be a bit slow. Unfortunately, our genome isn't Moby Dick. It's roughly the size of 6,000 Moby Dicks. Each index is going to be in just about six million locations. It can't possibly read all those at the speed we require.
Okay, so if we have too many items per index value, we just have to make our index larger. What if we do 10-mers, or even 20-mers? Well, 10-mers gives us 410 entries - call it a million. That means each index will be found in about 6,000 locations. Kind of a lot, but maybe doable. If we do 20-mers - well, 420 is 1,099,511,627,776. This is more on the lines of what we need in an index - it's well beyond the number of characters in the genome, so each index shouldn't show up in more than one or two locations. There's just one small problem: We now have six billion entries with twenty-character identifiers, and the space our index needs is now up to 120 gigabytes!
Maybe we could tweak and tune and find a sweet spot, but instead we'll try a different approach entirely to indexing. Next time.
Part I: A Million Dollars Up For Grabs
Part II: Analyzing DNA with BWA
Part III: Analyzing DNA Programmatically
Part IV: Indexing the Human Genome
Monday, May 13, 2013
Analyzing DNA programmatically
Last time I discussed using a program called BWA to try to determine if a given sequence was part of the human genome or not. It didn't seem to do a great job. How do we approach it programmatically?
Let's review the problem first. You have a machine that has analyzed some DNA, and it's given you some sequences it's found. Roughly 300,000 of them. Some with maybe only five characters, some with a few hundred. We call them "reads".
 Recall that a DNA sequence, from a programming standpoint, consists of any number of any of four characters: A,C,G, and T. A human genome, in the end, is just a DNA sequence. So if the human sequence was "AAGGTTTCC" and your sequence is "AGGT", bing! You've got a match.
Recall that a DNA sequence, from a programming standpoint, consists of any number of any of four characters: A,C,G, and T. A human genome, in the end, is just a DNA sequence. So if the human sequence was "AAGGTTTCC" and your sequence is "AGGT", bing! You've got a match.
Now, here's the catch: a read, instead of being four characters, is reasonably a minimum of fifty characters. The human genome, instead of being nine characters, is roughly six billion characters. 6,000,000,000 characters. If your first thought is to fire up a text editor and do a search, you had better make sure that (a) the editor is capable of handling a six gigabyte file, and (b) that your computer has six gigs available to throw around (In 2013, a high-end laptop will probably have eight gigs, so just barely enough space to hold the genome and run a couple of other programs.)
Another, minor issue is that most genome files are formatted with sixty-character lines. If your sequence is split across two lines, a simple text search won't find it. You can preprocess the file to remove all the carriage returns, but if you do, your text editor had better be able to handle one line with 6 billion characters on it!
Your next idea: grep. Will grep find a 50-character match in a genome? I couldn't find a good way to do it reliably, since, again, genome files tend to be formatted into 60-character lines, and grep isn't really capable of finding a string that breaks across a line. Moreover, grep is a line-based tool, so odds are if you try preprocessing the line, grep will think (rightly) that you have a file with one six-billion character line in it, try to read in the line, and run out of memory.
Here's the other catch: Suppose you could get grep to work, and it managed to search the whole genome in three seconds. But we have 300,000 reads to get through! That's about 250 hours worth of search time and we're under a three-hour deadline. So we can't really afford to search the entire genome for each read.
So we'll write a script. A good processing plan might be this: index the genome, index the reads, then march down the two together to find our list of best prefixes. Sounds great!
But it turns out that even indexing a file the size of the human genome is a pain. I'll think about why next time.
Also see: A Million Dollars Up For Grabs
Let's review the problem first. You have a machine that has analyzed some DNA, and it's given you some sequences it's found. Roughly 300,000 of them. Some with maybe only five characters, some with a few hundred. We call them "reads".
Now, here's the catch: a read, instead of being four characters, is reasonably a minimum of fifty characters. The human genome, instead of being nine characters, is roughly six billion characters. 6,000,000,000 characters. If your first thought is to fire up a text editor and do a search, you had better make sure that (a) the editor is capable of handling a six gigabyte file, and (b) that your computer has six gigs available to throw around (In 2013, a high-end laptop will probably have eight gigs, so just barely enough space to hold the genome and run a couple of other programs.)
Another, minor issue is that most genome files are formatted with sixty-character lines. If your sequence is split across two lines, a simple text search won't find it. You can preprocess the file to remove all the carriage returns, but if you do, your text editor had better be able to handle one line with 6 billion characters on it!
Your next idea: grep. Will grep find a 50-character match in a genome? I couldn't find a good way to do it reliably, since, again, genome files tend to be formatted into 60-character lines, and grep isn't really capable of finding a string that breaks across a line. Moreover, grep is a line-based tool, so odds are if you try preprocessing the line, grep will think (rightly) that you have a file with one six-billion character line in it, try to read in the line, and run out of memory.
Here's the other catch: Suppose you could get grep to work, and it managed to search the whole genome in three seconds. But we have 300,000 reads to get through! That's about 250 hours worth of search time and we're under a three-hour deadline. So we can't really afford to search the entire genome for each read.
So we'll write a script. A good processing plan might be this: index the genome, index the reads, then march down the two together to find our list of best prefixes. Sounds great!
But it turns out that even indexing a file the size of the human genome is a pain. I'll think about why next time.
Also see: A Million Dollars Up For Grabs
Saturday, March 23, 2013
Analyzing DNA with BWA
Last time I discussed a million-dollar challenge posted by Innocenture. The challenge is to analyze a series of DNA reads and determine the source. My professor suggested using a program named BWA, a Burrows-Wheeler Aligner. Burrows-Wheeler alignment is an algorithm for matching up two sequences which may not be the same length. Perhaps one sequence has two extra nucleotides in the middle but the entire rest of the sequences match. These kinds of sequences are very difficult to find naively, and which match is better can come down to a judgment call.
So the goal is to align each sequence in the example file to the human genome. BWA, for speed, asks that you index the target genome - the human genome, in this case - which takes a fair amount of time. Then, you pass it each sequence on the command line, with a command something like:
>bwa align AAGCTCTA human_genome
and it does its magic and returns the location in the genome of the best several matches.
So I worked up a Python script to analyze the input file and pass each sequence to BWA in that format, and let it run for a few hours. (I'm never sure how many because I never remember to change the machine's power settings to not switch itself off after a few minutes of no UI activity). But it eventually completed. I went to look at the results, and I found them very intriguiing. Although, according to the challenge, no less than 90% of the DNA was human, the BWA program only managed to match about 50%, about 150,000 reads.
I took a closer look at the BWA manual, and found this option:
If I understand this correctly, it means that if the alignment has more than one gap in it, BWA will discard it as not being a match. You can change the value of this parameter, but when I did, it seemed to slow down the analysis quite a bit. I didn't let it complete, but based on the portions I did run I suspect it would have gone over the three-hour limitation - at least on my workstation, which I'm sure is underpowered compared to the target hardware.
So I started to think about what sort of coding would need to be done to meet this challenge. Next time I'll think about Analyzing DNA Programmatically
So the goal is to align each sequence in the example file to the human genome. BWA, for speed, asks that you index the target genome - the human genome, in this case - which takes a fair amount of time. Then, you pass it each sequence on the command line, with a command something like:
>bwa align AAGCTCTA human_genome
and it does its magic and returns the location in the genome of the best several matches.
So I worked up a Python script to analyze the input file and pass each sequence to BWA in that format, and let it run for a few hours. (I'm never sure how many because I never remember to change the machine's power settings to not switch itself off after a few minutes of no UI activity). But it eventually completed. I went to look at the results, and I found them very intriguiing. Although, according to the challenge, no less than 90% of the DNA was human, the BWA program only managed to match about 50%, about 150,000 reads.
I took a closer look at the BWA manual, and found this option:
| -o INT | Maximum number of gap opens [1] |
If I understand this correctly, it means that if the alignment has more than one gap in it, BWA will discard it as not being a match. You can change the value of this parameter, but when I did, it seemed to slow down the analysis quite a bit. I didn't let it complete, but based on the portions I did run I suspect it would have gone over the three-hour limitation - at least on my workstation, which I'm sure is underpowered compared to the target hardware.
So I started to think about what sort of coding would need to be done to meet this challenge. Next time I'll think about Analyzing DNA Programmatically
Friday, March 22, 2013
A million dollars up for grabs
Of course, you have to do a few things to earn it. Innocenture is running a challenge, with a million dollar prize, for the ability to take a DNA read, analyze it, and determine the precise species of each read. Here's the kicker: it has to be done rapidly.
They have some examples available. The input is in a large XML file that contains the DNA reads and some information on the quality of the reads, and they provide you with the output file they would be looking for. One example has a little more that 300,000 reads of between 50 and 200 nucleotides, putatively taken from a human. According to the output, at least 90% of the reads are from human DNA.
So how do we duplicate this output file? There's a program called BLAST available on the web for bioinformatic analysis - you give it a sequence of DNA and it almost immediately comes back with the closest matches across their entire, huge DNA database.
So, we might be able to slam that database with the reads and get back the results. There's just one problem - notice I said 300,000 reads? Suppose we could get back each one in one tenth of a second. That makes 30,000 seconds, or a total of eight hours and 20 minutes of runtime. Sadly, the million dollars probably won't be given away unless you can get the reads done in under three hours. Oh, and did I mention the application won't actually have internet access?
So the BLAST site is out, which is unfortunate, because it really does an amazing job at matching sequences. What do we do instead?
Due to the nature of science in the United States - "publish or perish" - there are a whole lot of little bioinformatics applications around. Mostly, people will write one, publish a paper about it, and then forget about it. There's no point in maintaining it or going back and improving it since there's no chance of writing another paper about it unless you change the algorithms significantly.
Still, a few of these applications manage to have some shelf life. I'll look at Analyzing DNA with BWA next.
Also see:
Analyzing DNA Programmatically
They have some examples available. The input is in a large XML file that contains the DNA reads and some information on the quality of the reads, and they provide you with the output file they would be looking for. One example has a little more that 300,000 reads of between 50 and 200 nucleotides, putatively taken from a human. According to the output, at least 90% of the reads are from human DNA.
So how do we duplicate this output file? There's a program called BLAST available on the web for bioinformatic analysis - you give it a sequence of DNA and it almost immediately comes back with the closest matches across their entire, huge DNA database.
So, we might be able to slam that database with the reads and get back the results. There's just one problem - notice I said 300,000 reads? Suppose we could get back each one in one tenth of a second. That makes 30,000 seconds, or a total of eight hours and 20 minutes of runtime. Sadly, the million dollars probably won't be given away unless you can get the reads done in under three hours. Oh, and did I mention the application won't actually have internet access?
So the BLAST site is out, which is unfortunate, because it really does an amazing job at matching sequences. What do we do instead?
Due to the nature of science in the United States - "publish or perish" - there are a whole lot of little bioinformatics applications around. Mostly, people will write one, publish a paper about it, and then forget about it. There's no point in maintaining it or going back and improving it since there's no chance of writing another paper about it unless you change the algorithms significantly.
Still, a few of these applications manage to have some shelf life. I'll look at Analyzing DNA with BWA next.
Also see:
Analyzing DNA Programmatically
Thursday, March 21, 2013
RNA Polymerase ||| and the RIG-I pathway
A little story about immune responses in cells.
Type-I interferons (IFNs) are important for antiviral and autoimmune responses. They interfere with viruses as the viruses try to borrow the cell's replication mechanism to reproduce themselves.
The cell will produce interferons due to a couple of proteins: the retinoic acid induced gene I (RIG-I) and mitochondrial antiviral signaling (MAVS) proteins.
These, in turn, start the production process when cytosolic double-stranded RNA or single-stranded RNA containing 5′-triphosphate (5′-ppp) are nearby.
Here's a surprising thing: Cytosolic B-form double-stranded DNA can also induce IFN-β. For example, a DNA sequence of repeating AT can induce it (It’s known as poly(dA-dT). But no one knew how. Until a paper came out in 2009 by Yu-Hsin Chiu and a couple of other people. It turned out that inside the cell, the poly(dA-DT) was actually being converted into 5′-ppp.
But how? It turns out that an enzyme uses the poly(dA-dT) as a template to synthesizes 5′-ppp RNA. The enzyme is DNA-dependent RNA polymerase III (Pol-III). This was interesting because it was known that the Pol-III had a role in the nucleus of the cell, but not that it had to do with the immune system.
If you inhibit the working of Pol-III in a cell, and then introduce a bacteria like Legionella pneumophil, the bacteria grows in the cell. The implication is that Pol-III senses the DNA of the bacteria and triggers the IFN process.
Then, they put different things in the cell. Of all the things tested, only poly(dA-dT) activated the IRF3.
To ensure that there wasn’t something going on at another step in the path, some other things were tried: A silencing RNA strand was introduced into the cell that would stop the production of RIG-I and MAVS. No IFN-β was produced. DNASE-I is an enzyme that breaks down DNA. When that was introduced, no IFN-β was produced. On the other hand, IFN-β was produced in the presence of RNASE-I, so breaking down RNA had no effect.
Nucleic acids from the poly(dA-dT) cells were able to induce IFN- β, even in the presence of DNase I, so it wasn’t DNA that was causing it. Production stopped in the presence of RNase I though, so it must have been RNA that was being produced.
Similar tests were done to determine the exact length of the poly. As few as 30 base pairs were able to trigger the IFN. But, longer sequences with G’s and C have failed to trigger anything.
Type-I interferons (IFNs) are important for antiviral and autoimmune responses. They interfere with viruses as the viruses try to borrow the cell's replication mechanism to reproduce themselves.
The cell will produce interferons due to a couple of proteins: the retinoic acid induced gene I (RIG-I) and mitochondrial antiviral signaling (MAVS) proteins.
These, in turn, start the production process when cytosolic double-stranded RNA or single-stranded RNA containing 5′-triphosphate (5′-ppp) are nearby.
Here's a surprising thing: Cytosolic B-form double-stranded DNA can also induce IFN-β. For example, a DNA sequence of repeating AT can induce it (It’s known as poly(dA-dT). But no one knew how. Until a paper came out in 2009 by Yu-Hsin Chiu and a couple of other people. It turned out that inside the cell, the poly(dA-DT) was actually being converted into 5′-ppp.
But how? It turns out that an enzyme uses the poly(dA-dT) as a template to synthesizes 5′-ppp RNA. The enzyme is DNA-dependent RNA polymerase III (Pol-III). This was interesting because it was known that the Pol-III had a role in the nucleus of the cell, but not that it had to do with the immune system.
If you inhibit the working of Pol-III in a cell, and then introduce a bacteria like Legionella pneumophil, the bacteria grows in the cell. The implication is that Pol-III senses the DNA of the bacteria and triggers the IFN process.
How did they do it?
In a cell, they attached a luciferase reporter to the IFN-β promoter, so if the cell creates IFN-β, it would bioluminesce.Then, they put different things in the cell. Of all the things tested, only poly(dA-dT) activated the IRF3.
To ensure that there wasn’t something going on at another step in the path, some other things were tried: A silencing RNA strand was introduced into the cell that would stop the production of RIG-I and MAVS. No IFN-β was produced. DNASE-I is an enzyme that breaks down DNA. When that was introduced, no IFN-β was produced. On the other hand, IFN-β was produced in the presence of RNASE-I, so breaking down RNA had no effect.
Nucleic acids from the poly(dA-dT) cells were able to induce IFN- β, even in the presence of DNase I, so it wasn’t DNA that was causing it. Production stopped in the presence of RNase I though, so it must have been RNA that was being produced.
Similar tests were done to determine the exact length of the poly. As few as 30 base pairs were able to trigger the IFN. But, longer sequences with G’s and C have failed to trigger anything.
RNA Characteristics
Two enzymes, polynucleotide kinase (PNK) and shrimp alkaline phosphatase (SAP) are used by chemists: the former adds a phosphate group to a DNA or RNA molecule, the latter removes one. A third enzyme, Terminator Exonuclease, or Ter Ex, breaks apart RNA with exactly one phosphate at the 5’ end.
When the SAP was used to remove the phosphate, the RNA no longer induced IFN- β (the PNK had no effect). Even when the PNK was used to add back the phosphate that was removed, there was still no induction, implying that a single phosphate was inadequate. Similarly, treating the RNA with Ter Ex also made no difference.
Another pair of RNase enzymes break apart specifically single stranded RNA (ssRNA) or double stranded RNA (dsRNA). RNase III breaks apart dsRNA, while RNase T1 breaks apart ssRNA. RNase III turned out to inhibit the IFN- β, indicating that dsRNA was required.
When the SAP was used to remove the phosphate, the RNA no longer induced IFN- β (the PNK had no effect). Even when the PNK was used to add back the phosphate that was removed, there was still no induction, implying that a single phosphate was inadequate. Similarly, treating the RNA with Ter Ex also made no difference.
Another pair of RNase enzymes break apart specifically single stranded RNA (ssRNA) or double stranded RNA (dsRNA). RNase III breaks apart dsRNA, while RNase T1 breaks apart ssRNA. RNase III turned out to inhibit the IFN- β, indicating that dsRNA was required.
Put all these together and it seems that the trigger is dsRNA with multiple phosphate groups attached.
So the chain takes you from the poly(dA-dT) to a 5′-ppp.
Conclusion
Other tests bring you to the conclusion that Pol-III is the enzyme that triggers this conversion. Thus, Poly-III, in the cytoplasm of a cell, actually acts as a DNA sensor that will trigger an immune response. An entirely different function from the one it has inside the cell nucleus. Quite a surprise!
Friday, December 21, 2012
Unit Test Coverage in Biopython
This fall, for the first time, I've had the professional opportunity to work on some Python code. I find the language quite elegant and simple to write, much more so than Ruby, although I couldn't really say why. Along with my work on a Master's degree in bioinformatics, I decided to look at the Biopython project and see about adding some code coverage statistics to it.
Biopython is an open-source software project that provides Python libraries for a variety of bioinformatics purposes. It includes, among other features, a variety of file parsers, alignment algorithms, and interfaces with various common bioinformatics tools and databases. To assure quality, a battery of automated tests is run against the Biopython source code before every release. While Biopython has a substantial amount of automated tests, no statistics are gathered concerning the code coverage of the tests.
It took several steps. The first was simply to run the existing test suite; I downloaded the codebase to my Windows machine, and tried to build it, but it turned out to be really difficult. Biopython has a dependency on NumPy as well as several other packages. I shaved several yaks attempting to get it running, but gave up eventually. Instead, I created a virtual Linux machine and installed it on that.
This went much more smoothly. (Note that I don't find Linux to have any natural advantage here. People who play with the Biopython source code run it on Linux, so that install gets a lot more attention.) I was able to run the test suite after a fashion. Not all of the tests ran the first time, which is a bit scary - it's hard to modify code with which you're not familiar without passing tests. But I realized after a while that the suite does some things like connect to remote services to verify that the connections work, and those services were down. There's an option to run only tests that don't attempt connections, and that worked - again, after a fashion. There are so many external dependencies that the test suite is set up to skip tests that require packages that aren't installed. But as all I was looking for was a passing test suite, I was OK with that.
The next step was a coverage tool. I chose Ned Batchelder's Coverage which worked nicely. All I had to do was replace the line "python run_tests.py --offline" with "coverage run run_tests.py --offline" and the coverage ran. I found the architecture of the tool a little strange; you don't specify any kind of output format. Instead, the tool creates its own binary data file (prefixed with a '.' to hide it on Linux systems) and you run a separate command to generate output in various formats. But this worked well and I had my report, in a nice HTML format so I could look at it in a browser.
Coverage is nice, but I don't know that a single report is very useful. Comparing coverage between builds is where the true benefit lies. Are some tests no longer covering code that they were meant to? Were tests disabled for some reason? Why did the coverage percentage drop? Ideally, each time the code is built, a new coverage report would be generated. So, I went on to look at the Biopython build process.
Biopython uses Travis for its builds. Travis is a rather nice public continuous integration server that integrates with Github, where the Biopython source code lives. You provide a well-named build script in your source tree and Travis monitors your repository for changes. It was easy enough to incorporate the coverage tool, but Travis didn't provide a good mechanism for reporting. The simplest thing, I decided, was to use a script to automatically upload a file back to Github. I jumped through several more hoops to get this working - authentication, permissions - and today I find, first, that Github is deprecating file uploads and, second, that Travis is supporting a new artifact system . So I think most of that work is out the window.
Bother.
So I'll have to rework some of that. Nonetheless, the coverage effort gave me some interesting results. Biopython consists of 298 source files comprising 39,805 statements. Automated unit tests covered all but 12,499 of those statements, for a coverage percentage of 69%. 33 files (11%) had no coverage at all, while 60 files (20%) were fully covered. Based on these numbers, Biopython has at this moment an almost acceptable level of coverage, as it is a truism in the software development world that coverage rates above 70% tend to lead to diminishing returns of value.
However, as a code library, Biopython is free from the problems of automated user interface testing, which is one of the more difficult areas of automated testing. For this reason, one might expect a somewhat higher coverage rate. There is reason to suspect, however, that as time goes by, the percentage will go higher. An examination of some of the source files with zero coverage reveal deprecation warnings, i.e. indications to the user that the module in question should not be used for new development. At some point it is to be assumed that these files will no longer be part of Biopython, which will drive the code coverage percentage upwards.
The introduction of a code coverage measuring tool to the Biopython build process is an important step, but only a first one. The code coverage tool selected is particular to Python, while a certain amount of the Biopython source code is written in C, a much more difficult language to instrument even for automated unit testing, much less code coverage reporting on that testing. However, useful work could be done in attempting to add this coverage. The techniques used here only monitor statement counts – perhaps branching counts would be a valuable addition. Biopython also has many dependencies on third-party products, some of which are installed during unit testing and some are not. Tests around these integration packages might prove useful. Finally, some work could be done on analyzing differences in builds, sending out alerts to interested parties if a given build should have a sudden drop in the code coverage percentage, for example.
Code coverage is a useful tool for long-running projects. Hopefully some of this effort will make it into the main Biopython codebase!
Thursday, September 13, 2012
Denisovan Gene Sequencing
What the fossil record shows about Denisovans: They had at least one finger, one toe, and a tooth.
This evidence comes from the Denisova Cave in Siberia.Scientists found the finger, toe, and tooth, which came from three different individuals, in different levels of the cave, and after doing a DNA analysis determined that their last common ancestor with humans lived about a million years ago. (The fossils dated from about 50,000 years ago). Of course, with such a minimal amount of material available, it's a bit tricky to get a complete gene sequence. The fact that the cave is in Siberia helps some (the average temperature in the cave is right around freezing) but some nice work on sequencing from a group led by Matthias Meyer helped as well.
Here's what they did to the source material:
I'm sure you're kicking yourself for not thinking of it first. At any rate, the immobilization of the DNA on the beads seems to be the important part, as it allows for copying of the sequence thus creating extra source material to work with. We now know more about Denisovan gene seqences than we do about Neanderthal sequences, as the quality of these Denisovan genes is better, less contaminated, than anything we have from the Neanderthals. Pretty cool stuff! Here's an article from Ars Technica if you don't feel like wading through the original paper.
This evidence comes from the Denisova Cave in Siberia.Scientists found the finger, toe, and tooth, which came from three different individuals, in different levels of the cave, and after doing a DNA analysis determined that their last common ancestor with humans lived about a million years ago. (The fossils dated from about 50,000 years ago). Of course, with such a minimal amount of material available, it's a bit tricky to get a complete gene sequence. The fact that the cave is in Siberia helps some (the average temperature in the cave is right around freezing) but some nice work on sequencing from a group led by Matthias Meyer helped as well.
Here's what they did to the source material:
DNA is dephosphorylated, heat denatured, and ligated to a biotinylated adaptor oligonucleotide, which allows its immobilization on streptavidincoated beads.
I'm sure you're kicking yourself for not thinking of it first. At any rate, the immobilization of the DNA on the beads seems to be the important part, as it allows for copying of the sequence thus creating extra source material to work with. We now know more about Denisovan gene seqences than we do about Neanderthal sequences, as the quality of these Denisovan genes is better, less contaminated, than anything we have from the Neanderthals. Pretty cool stuff! Here's an article from Ars Technica if you don't feel like wading through the original paper.
Monday, September 03, 2012
Coding errors in DNA analysis software
The problem:
A method for analysing similarities in protein sequences is to use a substitution scoring matrix. The matrix will assign a specific score to each individual protein match, so you can compare the sequences by looking at each individual pair of proteins in the sequence, looking in the matrix to determine the compatibility score for the pair, and adding up (or otherwise aggregating) the total score. The higher the score, the more likely it is, presumably, that the two sequences are actually related.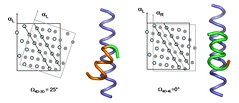
The art and science of comparing proteins
As a completely made-up example, if you have one sequence that goes Alanine - Glutamine - Isoleucine - Serine, and another sequence that goes Alanine - Glutamine - Tryptophan - Serine, you would look in the matrix for the scores for Alanine paired with Alanine, Glutamine with Glutamine, Isoleucine with Tryptophan, and Serine with Serine. If the identically matching pairs all scored 10, and the Isoleucine-Tryptophan pair scored 2, you would have a score of 32. On the other hand, if your matrix says that Isoleucine paired with Tryptophan gives you a -50, your score would total -20.
The program:
It's obviously pretty important to your analysis what matrix you use. Lots of people have come up with lots of matrices. One in particular, known as BLOSUM, was introduced in 1992 by Steven and Jorja Henikoff. The Henikoffs wrote a program (source code here) that analyzed a slew of already-matched sequences and assigned them scores, based on the probability that you would see them in a matching pair and the probability that you would see them at all. (This factor is relevant since some proteins overall appear with more frequency than others. You can find more details in this paper). They wrote it all up in a nice paper and the BLOSUM matrices have been in heavy use ever since.
But the funny thing is, there was a bug.
Yes, yes, I know what you're thinking: a bug? In a piece of software? Impossible! But it's true, and the bug wasn't pointed out, from the time the program was written in 1992, until 2008. It seems that, according to a paper by Mark Styczynski et al, there was an "incorrect normalization during a weighting procedure", which caused the program to generate different matrices based on the ordering of the sequences used as source.
So why did it take so long to find?
I'm thinking that the primary reason is because the source code is pretty incomprehensible. I'm not blaming the authors - I was writing code in 1992 and I'm sure that if I could even find any of it, it would be no easier to understand. My code, like theirs, would have been written in C, and in 1992 I think we were lucky to get code to work at all. Today, we have niceties such as unit testing, powerful IDE's, language features like garbage collection, and automatically generated documentation. In 1992, we had lint, and sometimes we even used it. Today, we have the confidence to make changes to the code strictly for reasons of readability and clarity. In 1992, we ran the program, looked at the output, and hoped it looked right. If it did, we were done. Plus, we had lint.
Here's the second reason the bug took so long to find: The matrix that was generated by the buggy code...worked, as far as anyone could see. People used the matrix for years and were perfectly satisfied with the results. As a matter of fact, Styczynski ran a comparison with the corrected matrices and determined that results were poorer using them than they had been using the buggy ones. I'm not sure why or how that could be. I suppose it might simply be that the Henikoffs happened to get lucky to create the better matrix, or tried a bunch of different ways to build a matrix and chose the most successful one to write about, not realizing that the paper described a slightly different algorithm than the code was actually running. Or even that the method Styczynski used to compare the matrices was wrong.
Whatever the reason, it's hard to argue with Styczynski's conclusion: "there is significant room for improvement in our understanding of protein evolution.".
But there's no question...
Whatever the reason, it's hard to argue with Styczynski's conclusion: "there is significant room for improvement in our understanding of protein evolution.".
Tuesday, August 21, 2012
On the teaching of genetics
Rosemary J. Redfield wrote an article on the teaching of genetics that resonated with me. Apparently the standard theory for teaching genetics is a sort of reprise of the history of genetics - you start with Mendel and dominant and recessive genes, move on to genes being on chromosomes, and slowly get on, if you're lucky, by the end of the course, to the molecular analysis of genes. The theory behind it is that the students will stand in Mendel's shoes and ask, "Well, why are some genes dominant", which will be answered by the next phase of the course, which will prompt more questions, to be answered by the following unit, and so on.
The problem is, that strategy doesn't work.
 It reminds me of astronomy classes both in high school and college. Now, astronomy is an awesome and fascinating subject. Go pick up any popular science magazine with an article on astronomy and just check out the language that they use: "Quasar". "Black Hole". "Dark Energy". "Strange Planet". It's like the whole subject was created just to appeal to teenagers. There is a podcast dedicated to astronomy called AstronomyCast that goes over a lot of this stuff, and my eleven-year-old son cannot go to sleep at night without listening to at least a few episodes.
It reminds me of astronomy classes both in high school and college. Now, astronomy is an awesome and fascinating subject. Go pick up any popular science magazine with an article on astronomy and just check out the language that they use: "Quasar". "Black Hole". "Dark Energy". "Strange Planet". It's like the whole subject was created just to appeal to teenagers. There is a podcast dedicated to astronomy called AstronomyCast that goes over a lot of this stuff, and my eleven-year-old son cannot go to sleep at night without listening to at least a few episodes.
But I hope that the interest isn't torn out of him in high school. If his courses are anything like mine, they will discuss: Stonehenge. Galileo. How, if you stay up night after night, you can see the position of the planets change slightly in relation to the stars. How an optical telescope works.
Genetics is just the same. Engage the student's interest by hitting them with the cool stuff first. Don't try to emulate the thinking of Mendel, because the instruments and techniques we use today are so much more powerful than Mendel ever dreamed of, and students know that, and often know what the techniques are. Redfield suggests starting with personal genomics, which seems like a good plan. Students, who know that they have a unique genetic makeup, should be interested in knowing what that makeup is, or at least how to find out. This would lead directly to the ethical questions surrounding that knowledge, and the course is off and running. Redfield is on to something.
The problem is, that strategy doesn't work.
It reminds me of astronomy classes both in high school and college. Now, astronomy is an awesome and fascinating subject. Go pick up any popular science magazine with an article on astronomy and just check out the language that they use: "Quasar". "Black Hole". "Dark Energy". "Strange Planet". It's like the whole subject was created just to appeal to teenagers. There is a podcast dedicated to astronomy called AstronomyCast that goes over a lot of this stuff, and my eleven-year-old son cannot go to sleep at night without listening to at least a few episodes.But I hope that the interest isn't torn out of him in high school. If his courses are anything like mine, they will discuss: Stonehenge. Galileo. How, if you stay up night after night, you can see the position of the planets change slightly in relation to the stars. How an optical telescope works.
“Students know what the cool stuff is.”
This isn't the cool stuff. Students know what the cool stuff is. If they don't listen to AstronomyCast, they've probably seen episodes of Nova, or at the very least, played a videogame in which black holes or wormholes play an integral part.Genetics is just the same. Engage the student's interest by hitting them with the cool stuff first. Don't try to emulate the thinking of Mendel, because the instruments and techniques we use today are so much more powerful than Mendel ever dreamed of, and students know that, and often know what the techniques are. Redfield suggests starting with personal genomics, which seems like a good plan. Students, who know that they have a unique genetic makeup, should be interested in knowing what that makeup is, or at least how to find out. This would lead directly to the ethical questions surrounding that knowledge, and the course is off and running. Redfield is on to something.
Saturday, August 18, 2012
DNA as storage mechanism
It seems some East Coast researchers are pushing the envelope in storing information in DNA. They encoded a book, roughly 5 megabits of data, into oligonucleotides, which were "synthesized by ink-jet printed, high-fidelity DNA microchips" which I don't fully understand but I presume means "made into a glob of DNA". The authors then sequenced the DNA and recovered all the data, with 10 incorrect bits.

The innate four bases (A,G,C,T) of DNA seem to lend themselves to some interesting storage techniques. The authors used simple redundancy for their storage - A and C both represented 0, G and T were 1, which was apparently a departure from earlier attempts which encoded each pair of bits into a single base. This made it easier to construct more robust sequences. I wonder if additional error-handling could have been done by placing checksum bases at intervals along the strand? Two bases would provide a range of 16 possible checksum values which seems it would handle a nice string of bits.
The book that was encoded had 50,000 words and eleven pictures. With an average code space of 40 bits per word, the text should have taken a tiny fraction of the total space, with the images providing the majority. Suppose that all ten bit errors were in one picture? It would be interesting to know how tightly compressed the images were. With a high compression factor, some of the bit errors might be substantial, but small changes to the compression might make a large difference in the visibility of any bit errors.
The authors say that DNA storage is dense, stable, and energy-efficient, but prohibitively expensive and slow to read and write compared to more standard storage. It will be fun to see how this technology evolves!
The innate four bases (A,G,C,T) of DNA seem to lend themselves to some interesting storage techniques. The authors used simple redundancy for their storage - A and C both represented 0, G and T were 1, which was apparently a departure from earlier attempts which encoded each pair of bits into a single base. This made it easier to construct more robust sequences. I wonder if additional error-handling could have been done by placing checksum bases at intervals along the strand? Two bases would provide a range of 16 possible checksum values which seems it would handle a nice string of bits.
The book that was encoded had 50,000 words and eleven pictures. With an average code space of 40 bits per word, the text should have taken a tiny fraction of the total space, with the images providing the majority. Suppose that all ten bit errors were in one picture? It would be interesting to know how tightly compressed the images were. With a high compression factor, some of the bit errors might be substantial, but small changes to the compression might make a large difference in the visibility of any bit errors.
The authors say that DNA storage is dense, stable, and energy-efficient, but prohibitively expensive and slow to read and write compared to more standard storage. It will be fun to see how this technology evolves!
Tuesday, July 03, 2012
Back to school!
In the fall I'll be enrolling for classes once more. Cathy's now finished her Doctor of Nursing Practice degree so now it's my turn. I've been accepted to the bioinformatics program at Indiana University. It appears that my first classes will be: Intro to Biology, Intro to Informatics, and Intro to Bioinformatics. It should be a lot of fun - I'm really looking forward to it.
Subscribe to:
Posts (Atom)